Methylation of Cyclohexane-1,3-dione at the C2-Position
SyntheticPage 813
DOI:
Submitted: December 4, 2016, published: May 7, 2017
Authors
Christopher Brown (brow3492@umn.edu)
Jessica Shudy (Jessica.Shudy@gmail.com)
Kenneth Tritch (trit0026@umn.edu)
Wayland Noland (nolan001@umn.edu)
A contribution from
Chemicals
synthesis:
Cyclohexane-1,3-dione (97%; Aldrich)
Acetone (ACS grade; Fisher Chemical)
Potassium carbonate (ACS grade; Fisher Chemical)
Methyl iodide (99%; Aldrich)
Sodium sulfate (anhydrous, 99%; Fisher Chemical)
Dichloromethane (ACS grade; Fisher Chemical)
Ethyl acetate (ACS grade; Fisher Chemical)
Hexane (ACS grade; Fisher Chemical)
TLC visualization:
(2,4-Dinitrophenyl)hydrazine (97%; Aldrich)
Ethanol (190 proof, ACS/USP grade; Pharmco-AAPER)
Sulfuric acid (95–98%; VWR Analytical)
Procedure
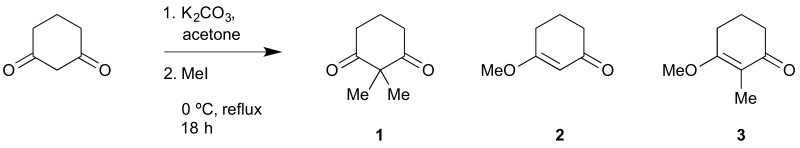
Cyclohexane-1,3-dione (5.00 g, 44.6 mmol, 1 eq.) was dissolved in acetone (100 mL) in a round-bottomed flask. Potassium carbonate (13.6 g, 2.2 eq.) was added, and then the resulting mixture was placed in an ice bath, and stirred for 15 min. Methyl iodide (6.1 mL, 2.2 eq.) was added. The resulting mixture was gradually warmed from 0 ºC to reflux over 2 h, and then was refluxed for 18 h more. The resulting cloudy, white mixture was allowed to cool to room temperature, and was then separated by suction filtration. The filter cake was washed with dichloromethane (25 mL). The filtrate was concentrated on a rotary evaporator, dissolved in water (50 mL) and dichloromethane (10 mL), and then extracted with dichloromethane (2 × 25 mL). The filter cake was placed in a conical flask with water (100 mL) and dichloromethane (50 mL). The resulting mixture was stirred for 10 min. The combined dichloromethane portions were washed with brine (25 mL), dried with sodium sulfate, filtered, and then concentrated on a rotary evaporator. The resulting yellow oil (6.08 g) was separated by column chromatography (1:8–1:1 ethyl acetate:hexane on SiO2). The desired fraction was concentrated on a rotary evaporator, and then dried under vacuum (0.1 mm Hg, room temperature, 2 h), giving a white powder (3.17 g).
Author Comments
This reaction shows no apparent sensitivity to air or ambient moisture.
Precautions should be taken to avoid any potential human exposure to methyl iodide, because of its toxicity and carcinogenicity (Ref. 1).
Warming the reaction mixture slowly provides a modest improvement in C- versus O-methylation selectivity.
Ethyl acetate can be used in place of dichloromethane. However, phase separation may take 5-10 minutes.
The initial reaction mixture could be concentrated without filtration and then partitioned into solvent and water, although problematic bumping was observed when the white solid was not removed prior to concentration.
Less polarizable electrophiles tend to favor O-alkylation (Ref. 2). Thus, the use of alkyl bromides or ester-derived alkylating agents (i.e., dimethyl sulfate or carbonate) is likely to give C,C-dialkylated ketones in low yields. However, these electrophiles may be good choices for O-alkylation-derived products with regard to yield, cost, and safety (Ref. 3).
Product (1) has no strong 254 nm (U.V. light) chromophores. It is advisable to use (2,4-dinitrophenyl)hydrazine (DNPH) solution to assist with thin-layer chromatography visualization (Ref. 4). Our visualization solution was prepared by dissolving DNPH (12 g) in ethanol (200 mL) and water (80 mL), followed by the careful addition of sulfuric acid (60 mL). Care should be taken not to allow solidified DNPH to accumulate, especially on surfaces that are exposed to friction, such as the threads of jar lids. Crystallized DNPH can be shock-sensitive.
No condition was found that could completely suppress O-methylation. From the given procedure, products (2) and (3) were generated in 37% and 9% yields, respectively (calculated by 1H NMR analysis of the extraction concentrate).
Data
Yield 51%.
M.p. 32–35 ºC (lit. 35–36 ºC; Ref. 5)
Rf 0.31 on SiO2 in 1:1 ethyl acetate:hexane
1H NMR (400 MHz, CDCl3) δ 2.67 (t, J = 6.9 Hz, 4H; C4, C6), 1.94 (p, J = 6.9 Hz, 2H; C5), 1.30 (s, 6H; CH3).
13C{1H} NMR (126 MHz, CDCl3) δ 210.5 (2C, C=O; C1, C3), 61.8 (1C, Cq; C2), 37.4 (2C, CH2; C4, C6), 22.3 (2C, CH3), 18.1 (1C, CH2; C5). Atom assignments are based on DEPT135 and HMBC analyses.
ESI-MS m/z calcd for [M+Na]+ 163.0730, found 163.0730.
IR (KBr, cm–1): ν(C–H) 2969, 2944, 2874; ν(C=O) 1728, 1697.
Lead Reference
Shibuya, S.; Isobe, M. Tetrahedron 1998, 54, 6677–6698.
Other References
1. Bolt, H. M.; Gansewendt, B. Crit. Rev. Toxicol. 1993, 23, 237–253.
2. Desai, R. D. J. Chem. Soc. 1932, 1079–1088.
3. Selva, M.; Perosa, A. Green Chem. 2008, 10, 457–464.
4. Leonard, J.; Lygo, B.; Procter, G. Advanced Practical Organic Chemistry, 2nd ed.; Chapman & Hall: Glasgow, G64 2NZ, UK, 1995, esp. pp 148–149.
5. De Jongh, H. A. P.; Wynberg, H. Tetrahedron 1965, 21, 515–533.
Keywords
alkylation, ketones, nucleophilic, substitution