Addition of a lithium acetylide to an aldehyde
SyntheticPage 137
DOI:
Submitted: August 24, 2001, published: August 24, 2001
Authors
Melanie Reich (m.t.reich@sussex.ac.uk)
A contribution from
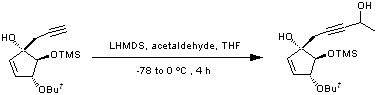
Chemicals
cyclopentene (prepared, 1 equiv.),
tetrahydrofuran (distilled from sodium-benzophenone ketyl, 5 mL/mmol),
LHMDS (Aldrich, 1 M solution in THF, 2.5 equiv.),
acetaldehyde (Lancaster, extremely volatile, 1.1 equiv.),
saturated ammonium chloride solution (5 mL/mmol),
diethyl ether (GPR, 2 x 10 mL/mmol),
water (10 mL/mmol),
ethyl acetate (GPR, 10 mL/mmol)
tetrahydrofuran (distilled from sodium-benzophenone ketyl, 5 mL/mmol),
LHMDS (Aldrich, 1 M solution in THF, 2.5 equiv.),
acetaldehyde (Lancaster, extremely volatile, 1.1 equiv.),
saturated ammonium chloride solution (5 mL/mmol),
diethyl ether (GPR, 2 x 10 mL/mmol),
water (10 mL/mmol),
ethyl acetate (GPR, 10 mL/mmol)
Procedure
A solution of the cyclopentenol (344 mg, 1.22 mmol, 1 equiv.), in distilled tetrahydrofuran (6 mL), was cooled to -78 ºC and lithium hexamethyldisilazane (LHMDS) (3.1 mL of a 1 M solution in tetrahydrofuran, 3.05 mmol, 2.5 equiv.) was added. The mixture was allowed to stir for 2 hours keeping the temperature constant at -78 ºC; acetaldehyde (0.084 mL, 59 mg, 1.34 mmol, 1.1 equiv.) was then added. The reaction mixture was allowed to warm to 0 ºC over 2 hours, after which the reaction was quenched with saturated ammonium chloride solution (6 mL). Extraction with diethyl ether (2 x 12 mL) was followed by washing of the organics with water (12 mL) and re-extraction of the combined aqueous layers with ethyl acetate (12 mL/mmol). The combined organic fractions were dried over magnesium sulfate and the solvent reduced in vacuo. The crude oil was purified by flash column chromatography (petroleum ether/diethyl ether, 10 : 1 to 5 : 1) to yield the product as a pale yellow solid (185 mg, 46 % (92 % based on recovered sm)). Also obtained was starting material (172 mg, 50 %).
Author Comments
This reaction was carried out numerous times, on 100 to 500 mg of starting material, however only moderate yields could be achieved. Further improvement (e.g. use of different bases (KHMDS, etc.)) was not possible and as starting material was reclaimed from the reaction to give near quantitative recovery the yield was satisfactory.
Data
1 H NMR (300 MHz; CDCl3) 5.90 (1 H, dd, J 6.0 and 1.0, CH), 5.69 (1 H, dd, J 6.0 and 1.0, CH), 5.47 (1 H, q, J 6.5, CCHO), 4.16 (1 H, dt, J 5.5 and 1.0, CHO), 3.91 (1 H, d, J 5.5, CHO), 3.40 (1 H, s, OH [exch]), 3.08 (1 H, s, OH [exch]), 2.56 (1 H, dd, J 17.0 and 2.0, HCH), 2.19 (1 H, dd, J 17.0 and 2.0, HCH), 1.39 (3 H, d, J 6.5, CH3), 1.18 (9 H, s, (CH3)3C), 0.15 (9 H, s, (CH3)3Si)
Lead Reference
S. Caddick, S. Khan, N. J. Smith, D. M. Barr, V. M. Delisser, Tetrahedron Lett., 1997, 38, 5053